Software
![]()
Example: 3D Interpolation of Spatial Diversity using R. This is simply a demo of some various R functions for displaying a 2D surface (in this case genetic diversity values interpolated across space) in 3D format.
–
–
–
![]()
fasta2genotype.py – A python script that converts fasta output files from STACKS into a multitude of formats. Data can be output as a matrix of either sequences, SNPs, or haplotypes. Right now, formats include (1) migrate-n, (2) Arlequin, (3) DIYabc, (4) LFMM, (5) Phylip, (6) G-Phocs, or (7) Treemix. Additionally (8), the data can be coded as unique integers (haplotypes) in Structure, Genepop, SamBada, Bayescan, Arlequin, or GenAlEx formats, or as allele frequencies by population. Download the user manual here.
–
–
–
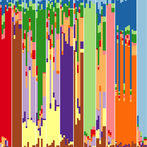
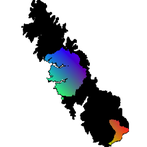
The following two R tutorials were written for Biology 624: Population Genetics (a graduate course taught by Dr. Andy Bohonak at SDSU). Both relate to population genomics and its applications. The first explores a ddRADseq dataset and compares it to a microsatellite dataset. The second identifies outlier loci and uses them to predict local adaptation under a future climate scenario. You can download data for the tutorial here.
–
–
Part A — Analysis of Population Structure: Microsatellites vs. ddRADseq
–
–
–
–
 –
–
–
Part B — Population Genomics: Identifying Outlier Loci and Predicting Future Adaptation
–
–
–
–
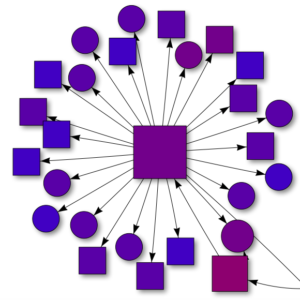
A network showing the academic pedigree for herpetology in California. Each connection shows descent, from adviser or mentor to student, and each shape is a student’s dissertation (master’s thesis or other in a few cases). The colors represent date of dissertation, from old (red) to recent (blue). The shapes represent taxon (amphibians, reptiles, both, or other). Since students can have more than one adviser, I chose the single most important one, generally the PhD adviser when applicable.

A more efficient way to run GATK4’s HaplotypeCaller | GenotypeGVCFs pipeline for de novo RNAseq SNP data. This pipeline is designed to: (1) Call SNPs in a pooled transcriptome, (2) Genotype (efficiently) the same SNPs in each sample, (3) Annotate the SNPs for effects such as synonymous/missense, etc., (4) Merge genotypes, SNP annotations, and gene annotations from a trinotate report
More soon!